集产品研发、生产、销售于一体
多年产品研发经验,自主知识产权
趣购彩18621315298
400-6885-928

集产品研发、生产、销售于一体
多年产品研发经验,自主知识产权
趣购彩18621315298
400-6885-928

第一作者:张广吉
通讯作者:王立强副教授,刘又年教授
通讯单位:中南大学化学化工学院,郑州大学材料科学与工程学院
论文DOI:10.1021/acscatal.2c01113
全文速览
开发可替代贵金属基催化剂、实现硝基化合物的高效加氢还原制备相应氨基化合物的非贵金属催化体系具有重要意义但仍充满挑战。该工作报道了一种多级多孔碳载N,S共配位稳定的钴单原子催化剂(Co1/NSC-AT),以实现硝基化合物高效加氢还原。Co1中心独特的配位环境结合碳载体的多级孔结构,赋予了Co1/NSC-AT优异的催化性能;加氢反应可以在温和的条件(35 oC, ~1 bar H2)下高效进行。理论计算表明,Co1/NSC-AT中的N,S共配位的钴单原子中心(Co1S1N3)是氢化反应的活性中心。与Co1N4和Co纳米颗粒相比,Co1S1N3具有较低的反应势垒。此外,N、S共配位环境可以调整Co单原子的电子结构,促进H从活性位点上的解吸,从而促进加氢过程。
背景介绍
硝基化合物还原是制备氨基化合物及其衍生物的重要手段。以贵金属为催化剂、氢气为还原剂的催化加氢技术是当前硝基还原的使用最为广泛的手段。然而,贵金属基催化剂存在成本高、加氢选择性差且易中毒等问题,限制了其大规模的推广使用。因此,发展高效非贵金属加氢催化剂是当前研究的趋势。杂原子掺杂碳负载的Fe 系(Fe、Co 和 Ni)金属,尤其是金属中心具有原子分散特征的碳负载铁系单原子催化剂,因其极低的成本、高选择性和良好的催化活性,是最有前景的催化剂之一。目前,铁系金属催化剂面临的挑战是如何将碳负载单原子催化剂的活性提高至贵金属基催化剂所具备的水平。该方面的研究虽然取得了一些研究进展,但单原子催化剂在硝基化合物的加氢反应中仍处于起步阶段,其催化性能仍有极大的提升空间。
研究出发点
单原子催化剂的活性受其金属中心的电子结构和空间分布影响。金属中心的电子结构主要受配位原子影响,其对反应物的吸附/解吸、中间体的形成以及产物的解离都具有重要的影响。然而,当前报道的碳负载单原子催化剂的金属中心多由N原子稳定。N原子电负性高,导致电子金属中心呈现缺电子状态,这对于反应物或中间体的吸附/解离并非总是优选的。另一方,并非所有单原子位点都是有催化活性的。单原子位点通常位于微孔结构中,仅有暴露在催化剂“外表面”的位点才是活性的。就此而言,介孔结构可以通过联通微孔,而使活性充分暴露。为此,作者提出了一种利用蛋白-金属离子网络化学策略,构建多级孔碳负载的、N, S共配位钴单原子催化剂,即Co1/NSC-AT。其兼具高效的反应位点Co1S1N3和多级孔结构带来的高的活性位点利用率。
图文解析
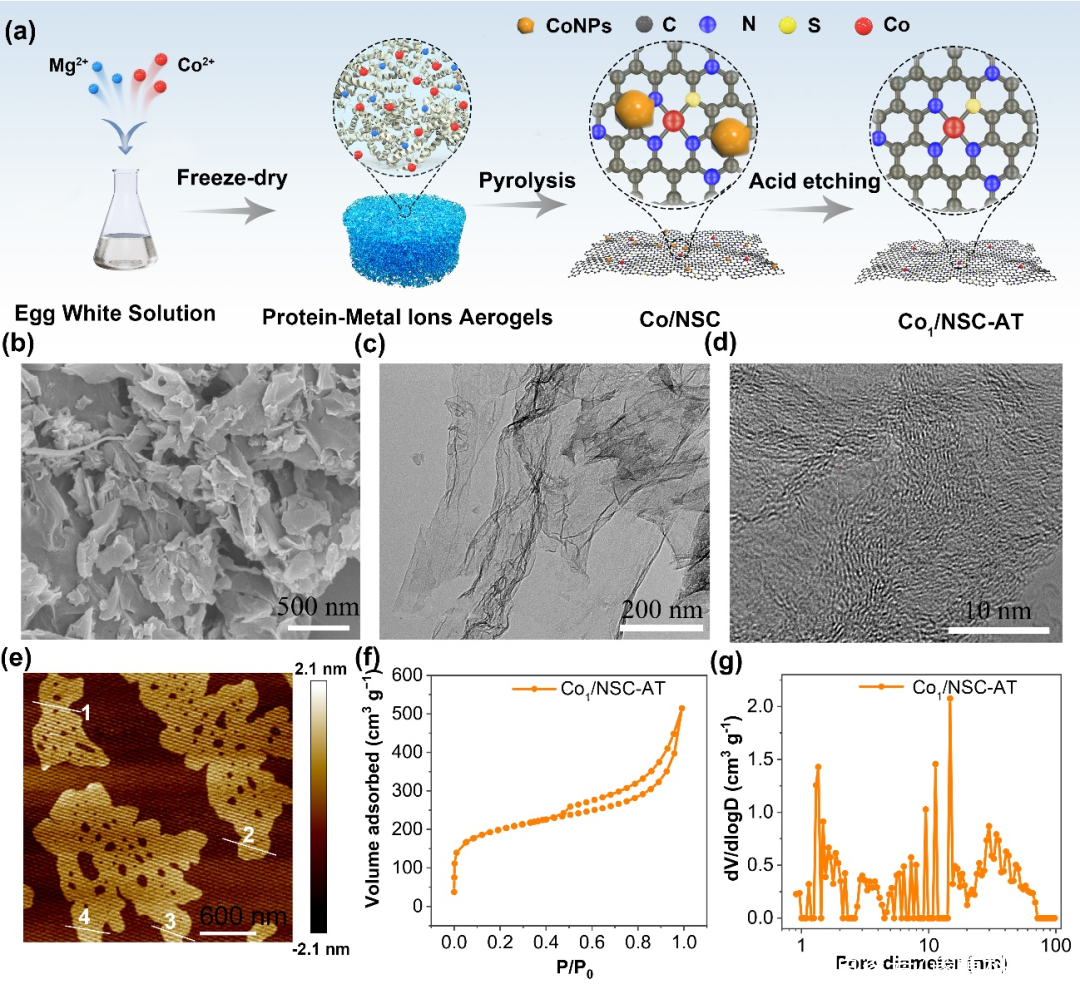
图1. 催化剂Co1/NSC-AT的(a)制备示意图.(b–e)扫描电镜、透射电镜、HR-透射电镜和原子力显微镜图像.(f) N2吸附等温线和(g)孔径分布.
Co1/NSC-AT的SEM,TEM和高分辨TEM分别如图所示。Co1/NSC-AT呈现出几何片状结构,由弯曲的多孔超薄石墨烯构成,AFM测试结果证明样品厚度有1.5~2.0 nm ,呈单层或少层石墨烯样。Co1/NSC-AT显示出多级孔结构,高的孔体积,大的BET比表面积。
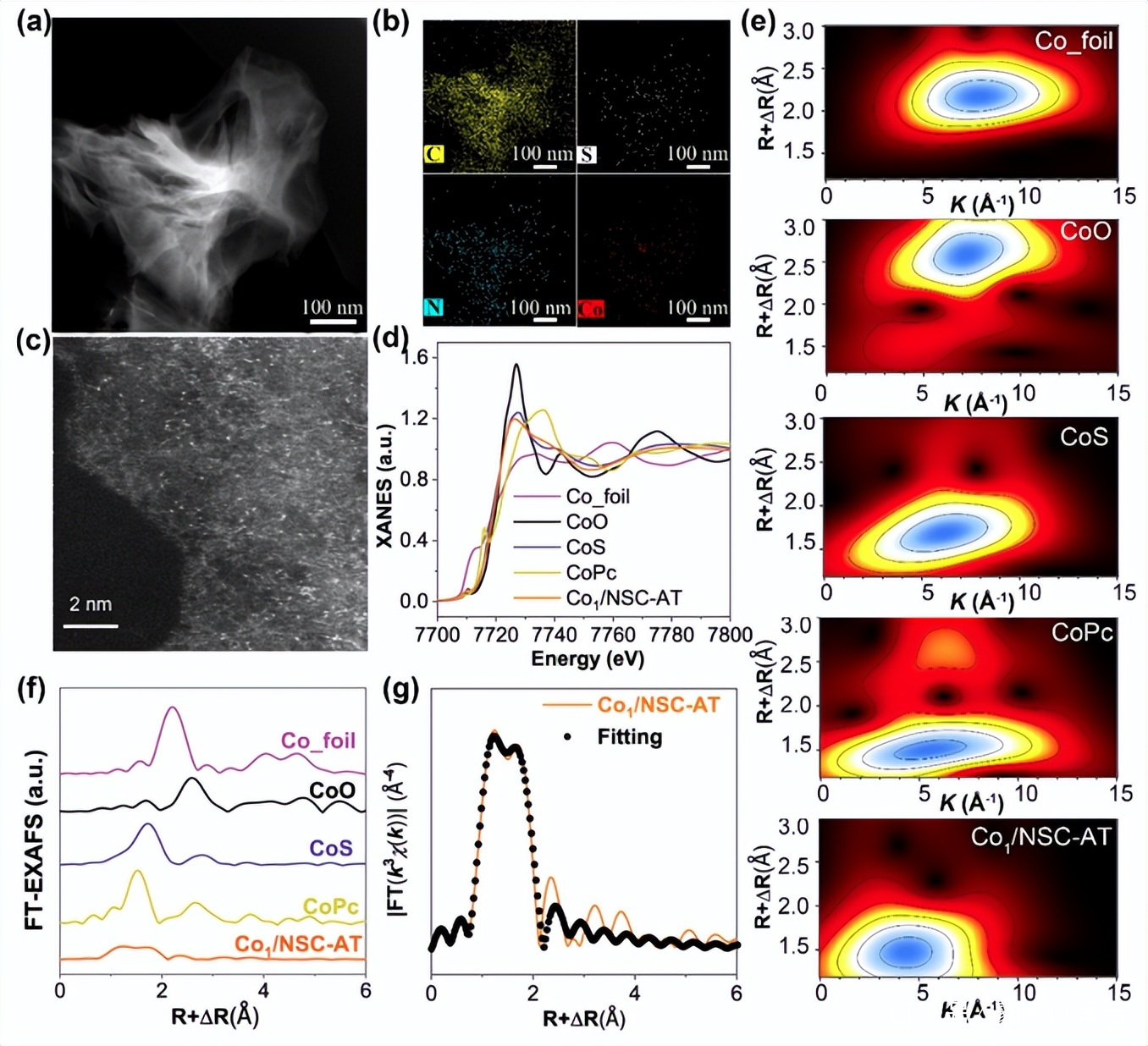
图2. Co1/NSC-AT的(a)HAADF-STEM图像和 (b) C、S、N 和 Co 的相应 EDX 元素映射 。(c)相差校正HAADF-STEM 图像。(d) K 边缘的 XANES 光谱。(e) 小波变换。(f) FT R空间曲线。(g)EXAFS的R空间拟合曲线。
Co1/NSC-AT的HAADF-STEM和相应的EDX mapping图像显示,Co,C,N和S均一的分布在碳基质中,而且金属钴以单原子形式存在,未发现钴纳米粒子。为了进一步探索Co原子的化学价态和配位环境,对Co1/NSC-AT进行了X射线吸收近边结构(XANES)和扩展X射线吸收精细结构光谱测量。结果表明Co1/NSC-AT中的Co带有正电荷,价态为~ +2。Co的 k边傅里叶变换k3加权EXAFS光谱和EXAFS小波转换分析可知,Co是单原子分散的,且被N、 S共配位稳定。EXAFS拟合计算出Co原子的配位数为4,N、S原子的比例为3:1,因此Co中心的理论配位结构为Co1S1N3,是催化剂Co1/NSC–AT中的主要催化活性位点。
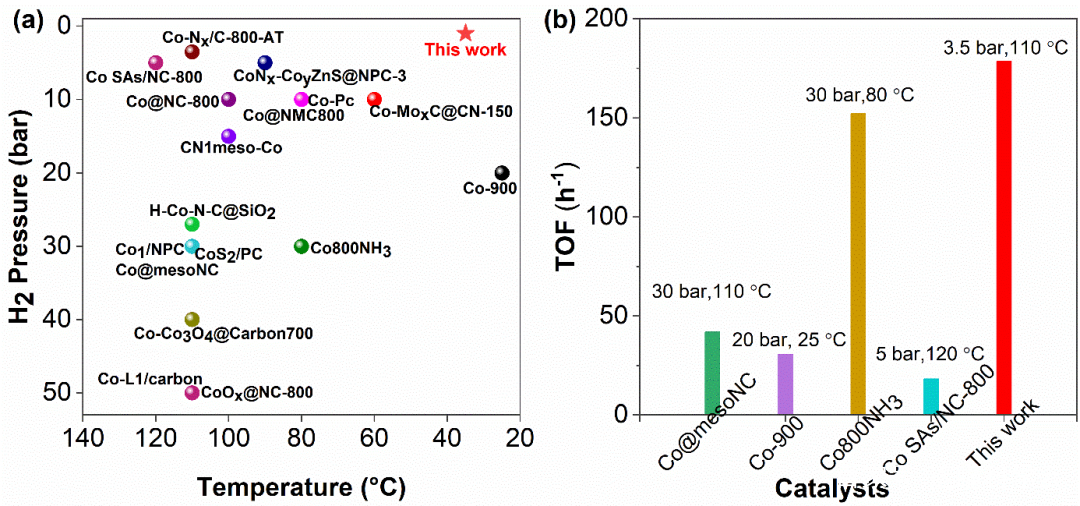
图3 催化剂Co1/NSC-AT与近来文献报道的钴催化剂硝基加氢性能对比
为了考察催化剂Co1/NSC-AT对不同硝基底物的催化选择性和普适性,进行了底物扩展实验(45个底物)。大部分的硝基加氢反应都可以在温和的条件下 (85 oC,5 bar)进行,转化率达到99%,选择性大于99%,优于最近报道的N配位稳定的钴单原子催化剂。
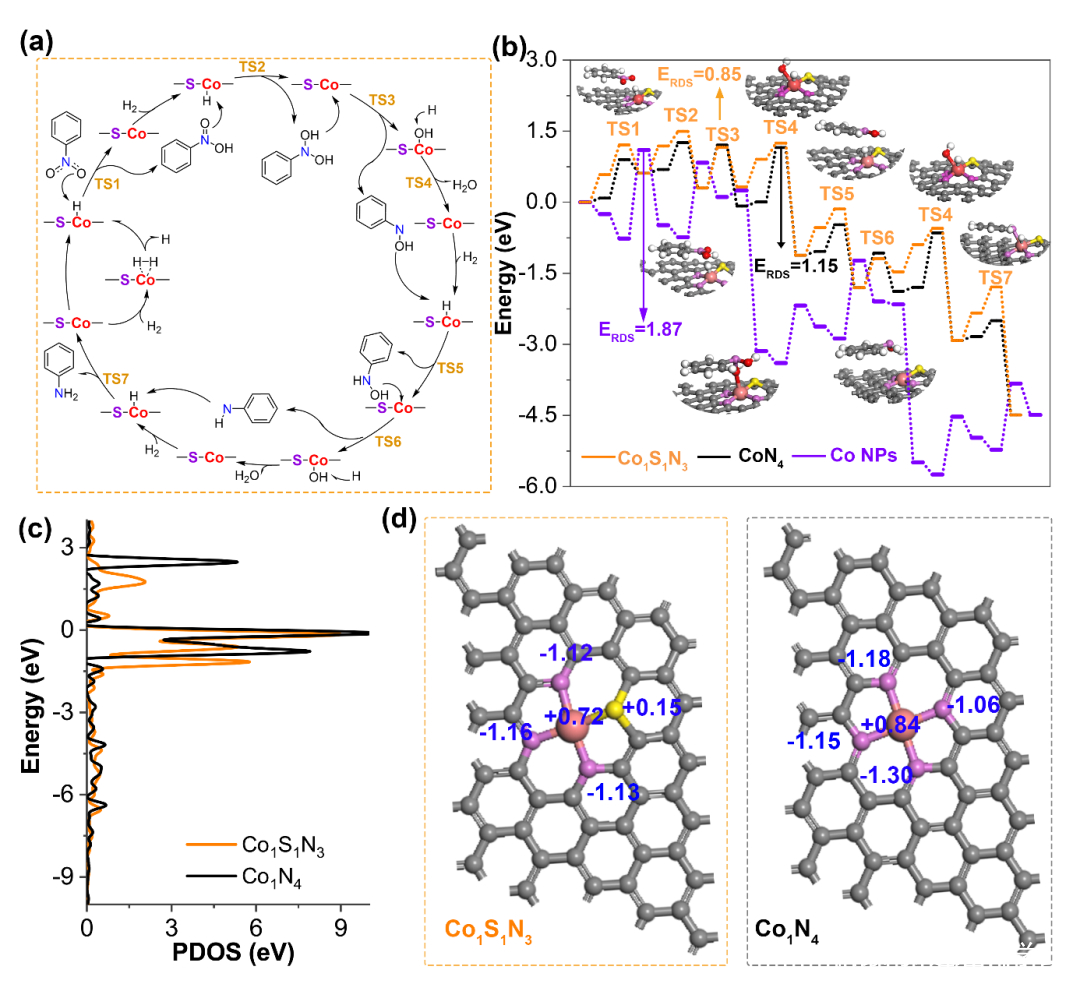
图4. (a) 硝基苯 (PhNO2) 在Co1S1N3 位点加氢生成苯胺 (PhNH2)的反应途径。(b) 在不同位点Co1S1N3,Co NPs和 Co1N4上硝基苯加氢的能量分布;(c) CoN4 和 Co1S1N3模型的DFT 计算预测状态密度;(d) 不同Co中心的Bader电荷.
为了研究硝基化合物在单原子Co1S1N3位点的催化机理,对硝基苯的催化加氢进行了DFT 计算模拟。在Co1S1N3位点上对硝基苯的加氢催化遵循直接途径,从C6H5NO2H2 到 C6H5NOH这一步在整个加氢反应过程中拥有最高的能垒 (0.85 eV),是反应的决速步骤。随后,通过局部态密度 (PDOS) 和Bader电荷分析,揭示了配位环境对与其催化性能密切相关的电子结构的影响。在Co1S1N3中Co 3d轨道的PDOS与Co1N4 中的相比能量水平低,表明Co1S1N3与H*的结合能力较弱,有利于H的脱附,促进加氢反应。由于Bader电荷分析可知, Co1N4 和 Co1S1N3位点上Co原子的电荷密度分别为 +0.72 和 +0.84 e。这表明S的引入可以降低Co 的氧化态,导致其与 H 的相互作用较弱。总体而言,N、S 配位结构为单原子中心提供了独特的电子结构,有利于硝基苯加氢反应的进行。
总结与展望
利用蛋白-金属离子网络构筑了多级孔碳负载的,N、S共配位稳定Co单原子催化剂,实现了硝基化合物的高活性、高选择性加氢。研究结果将为碳负载单原子催化剂的结构设计及性能优化、高性能硝基加氢催化剂的构建提供思路。
通讯作者介绍
刘又年,中南大学化学化工学院教授,博士生导师。获得省部级自然科学奖、科技进步奖等5项。多年来主要从事多功能纳米材料的制备及其在生物医用、药物控释、环境能源和光电热催化中的应用。在Adv. Mater., Angew. Chem. Int. Ed., PNAS, Adv. Func. Mater., ACS Nano, AIChE J等刊物发表学术论文200余篇,他引7000余次,H-Index 42,授权国家发明专利20余项。
王立强,郑州大学材料科学与工程学院副教授,硕士生导师。主要从事催化剂设计、铝资源的高效利用等方面的研究。在ACS Catal. Adv. Mater., Angew. Chem. Int. Ed., ACS Nano, 等刊物发表学术论文40余篇,他引2000余次。担任Journal of Central South University、Frontiers in Chemistry等杂志的青年编委或客座编委。
文献来源
Guangji Zhang, Feiying Tang, Xin Wang, Liqiang Wang, and You-NianLiu. Atomically Dispersed Co–S–N Active Sites Anchored on Hierarchically Porous Carbon for Efficient Catalytic Hydrogenation of Nitro Compounds. ACS Catalysis,2022, 12, 5786–5794.
原文链接:
https://pubs.acs.org/doi/full/10.1021/acscatal.2c01113.
浏览次数:513
浏览次数:592
浏览次数:794
浏览次数:793
浏览次数:1953
